An industrial placement at ISIS opens the door to numerous opportunities, from learning to doing and publishing science. This has also been the case for Matei Pascariu, a former placement student in the Molecular Spectroscopy Group. A recent collaborative effort involving Matei as first author, under the supervision of Dr Svemir Rudic and in collaboration with Professor Leonardo Bernasconi (University of Pittsburgh), Dr Matthew Krzystyniak (ISIS), and Dr James Taylor (ISIS) has delved into the complex world of furanosides to disentangle their complicated vibrational signatures. Their comprehensive study combines multiple spectroscopic techniques and theoretical approaches to shed light on this molecule's vibrational dynamics and structural properties.
Furanosides are similar to standard sugar molecules, but with a five-membered ring at their centre rather than one with six carbon atoms. They have great biological significance, with some examples forming parts of RNA and DNA. Understanding the structural and dynamical properties of furanosides is crucial as they play a vital role in various natural compounds and biochemical processes.
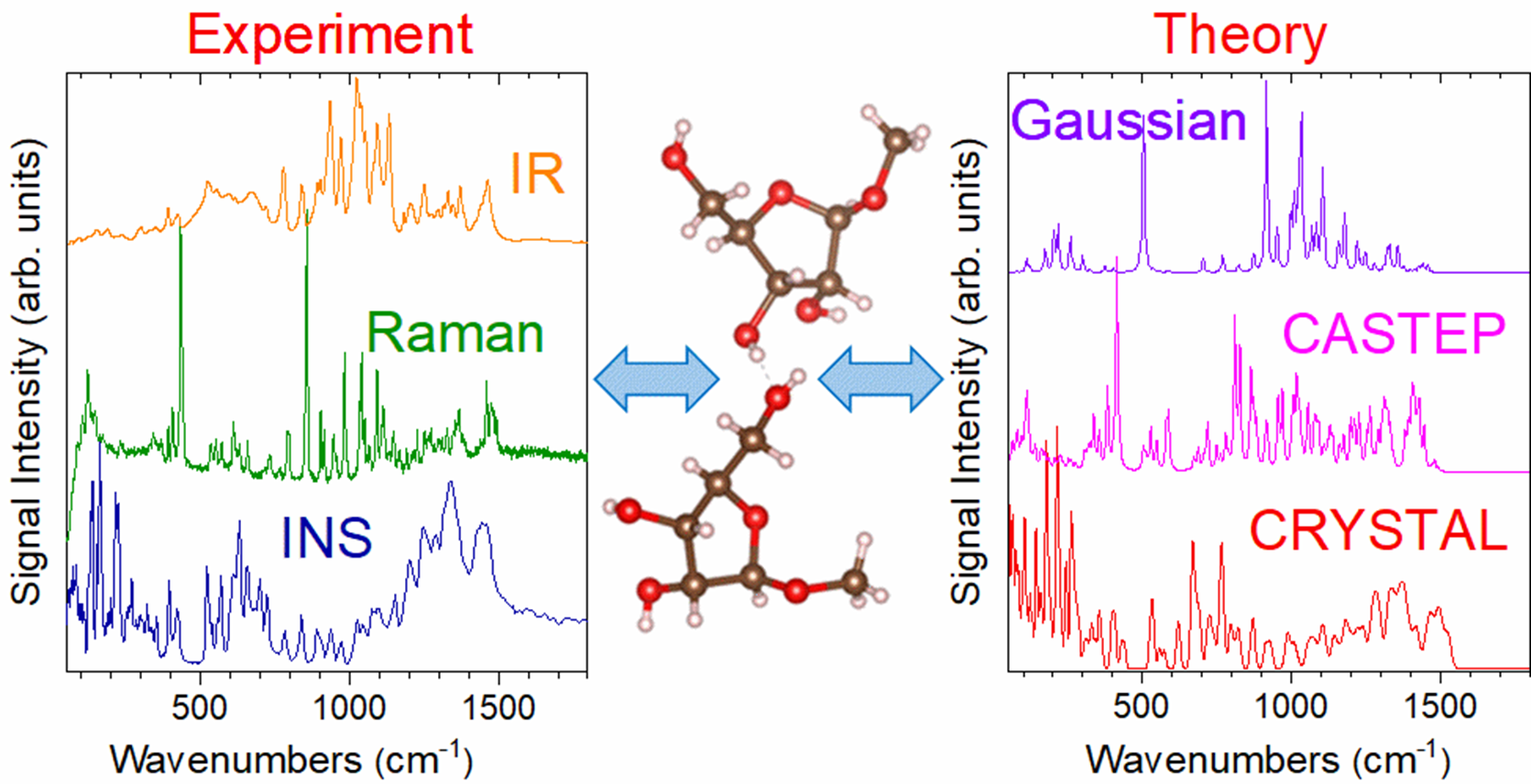
In this study, published in the Journal of Physical Chemistry A, the team utilized inelastic neutron scattering measured on TOSCA and Raman and infrared spectroscopy. These techniques allowed them to observe all vibrational modes of methyl-β-D-ribofuranoside, irrespective of selection rules. To complement these experimental methods, density functional theory (DFT) computational methods were employed, using both gas-phase (Gaussian) and solid-state (CRYSTAL, CASTEP) approaches. This dual approach represented an original effort to combine the clarity of single-molecule simulations with the complexity of the condensed-phase simulations to better understand the molecule's defining vibrational features.
Matei and the rest of the group identified distinct signatures from the two structures of methyl-β-D-ribofuranoside within the unit cell which are primarily differentiated by the orientation of the furanose ring O-H bonds. They detailed the vibrational spectra in various energy regions: low-energy (<400 cm-1), dominated by lattice vibrations and rotations of functional groups; mid-energy (400-900 cm-1), characterized by out-of-plane bending motions of the furanose ring; and high-energy (>2800 cm-1), encompassing the C-H and O-H stretching modes, offering convincing evidence of at least one hydrogen-bonding interaction between the two structures.
“The combination of experimental spectroscopic techniques with DFT calculations allowed us to explore the intricate behaviour of methyl-β-D-ribofuranoside more comprehensively than ever before," explains Matei. “This multifaceted approach not only provided detailed insights into the vibrational dynamics but also bridged significant gaps in the existing literature on this molecule."
This study proposes a rather different approach for future research, suggesting that combining multiple spectroscopic techniques with advanced computational methods can yield unprecedented insights into complex molecular systems.
![]()
The full paper can be found online at DOI: 10.1021/acs.jpca.4c00266